More Light Field Micrographs (2008)
Moving the sun around a human hair
April 25, 2008
Light field illuminator design: Marc Levoy and Ian McDowall
Photography, processing, web page: Marc Levoy
As an extension of our work on the light field microscope (LFM), we have
inserted a video projector and microlens array into the illumination path of an
optical microscope. This allows us to digitally control the spatial and
angular distribution of light (i.e. the 4D light field) arriving at the
microscope's object plane. We call this new device a light field illuminator
(LFI). Our prototype, which combines the LFM and LFI, is pictured at left
below. As in our light field microscope, diffraction places a limit on the
product of spatial and angular bandwidth in these synthetic light fields.
Despite this limit, we can exercise substantial control over the quality of
light incident on a specimen. In the experiment summarized at right below, we
demonstrate angular control over illumination, using a human hair as the
specimen.
Examining the photographs in the bottom row above, notice the enhanced
visibility of scattering inside the hair fiber under quasi-darkfield
illumination (2nd column) as compared to brightfield (1st column). Under
headlamp illumination (3rd column) note the specular highlight, whose
undulations arise from scales on the fiber surface. The view under oblique
illumination (4th column) is particularly interesting. Some of the light
reflects from the top surface of the hair fiber, producing a specular
highlight, but some of it continues through the fiber, taking on a yellow cast
due to selective absorption by the hair pigment. Eventually this light
reflects from the back inside surface of the fiber, producing a second
highlight (white arrow), which is colored and at a different angle than the
first. This accounts for the double highlight characteristic of blond-haired
people. For a more detailed explanation of this phenomenon, see Steve
Marschner et al., "Light Scattering from Human Hair Fibers", ACM
Transactions on Graphics (Proc. SIGGRAPH), Vol. 22, No. 3, pp. 780-791,
2003.


|
The MP4 movies at left shown the effect of changing the azimuthal direction of
oblique illumination, while holding the angle of declination constant at about
25 degrees relative to the optical axis. We call this protocol "moving the sun
around", since it simulates the path taken by the sun over a 24-hour period, if
you happen to be standing at the north pole on the day of the summer solstice.
The first movie shows the pattern displayed on our video projector, captured in
real time. (The brighter dot, which appears to circle the scene
counter-clockwise, was added to provide feedback, visible through the
microscope, of the current compass direction of the illumination.) The second
movie, captured in synchrony with the first, shows the effect of this changing
illumination on the single blond human hair. Note how the specular highlights
move, lending a sense of solidity to the hair fiber, as well providing shape
cues about the scales on its surface.
These movies are designed to be played in an
infinite loop, if your movie player supports this functionality.
|
Functional Neural Imaging of Larval Zebrafish
July 25, 2008 (and July 3, 2009)
Optical assembly: Ruben Portugues and Marc Levoy
Specimen and photography: Ruben Portugues
Processing and web page: Marc Levoy
We have helped several colleagues outside Stanford install light field
microscopes in their laboratories. One example is Professor
Florian Engert, of the Department of Molecular and Cellular Biology at
Harvard University. Prof. Engert studies the behaviorial, physiological, and
molecular mechanisms underlying synaptic plasticity. One of his primary model
systems is the larval zebrafish. With the help of his postdoctoral student
Ruben Portugues, we assembled a light field microscope on a vertical rail
system in his laboratory, and we looked at fluorescently labeled zebrafish.
Harvard University
Light Field Microscope

(Click for larger view.)
Photographed July 3, 2009.
|
Eye of larval zebrafish
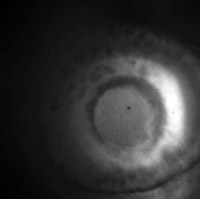
(Click to view
MP4 movie.)
Fluorescence image of the eye of a zebrafish 4 days post fertilization.
The fish belongs to a trangenic line that expresses green fluorescent protein
under the Ath5 promoter. This results in most of the retinal ganglion cells at
this age being labeled.
The fish is alive, mounted in low melting point agar on its side, so the view
is straight down into his eye, along the axis of the lens, which creates
visible optical distortion.
Click here for the
full-res light field.
Captured July 3, 2009.
|
Head of larval zebrafish
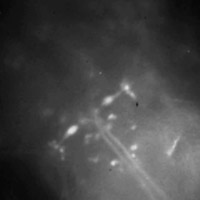
(Click for
MOV or
MP4.)
Live larval zebrafish at 6 days post fertilization, embedded in agar. The fish
was spinally injected with calcium green dye 24 hours in advance, which results
in a subset of all spinal projection neurons being labeled.
Many neurons are individually identifiable, most notably the two large
elongated Mauthner (M) neurons (one on each side) whose large axons cross the
midline and then project down the spinal cord.
Click here for the
full-res light field.
Captured July 25, 2008.
|
Functional neural imaging
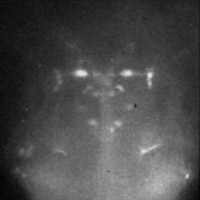
(Click for
MOV or
MP4.)
In this movie the fish was electroshocked every 5 seconds. This causes an
increase in fluorescence in the M neurons, which correlates with calcium influx
into these neurons and increased neuronal activity. The M neurons are known to
mediate the fast escape response: an aversive stimulus on the left side will
cause the left M neuron to fire and will result in contraction of the muscles
on the right side of the tail: the fish will therefore bend away from the
stimulus.
This video light field is too large to download.
Captured July 25, 2008.
|
The picture at left is of a light field microscope assembled in Florian
Engert's laboratory in about 2 hours on July 3, 2009. It is almost identical
to the device assembled on July 25, 2008. From top to bottom on the vertical
rail are a Retiga 4000R B&W camera (A) with a 2K x 2K pixel cooled sensor, two
Nikon 50mm f/1.4 lenses (B) placed nose-to-nose to form a 1:1 relay lens
system, a custom telescoping housing (C) containing a microlens array
(125-micron x 125-micron square lenses, focal length=2.5mm), a tube lens (D),
fluorescent illuminator (E) and Olympus 40x/0.8NA water dipping objective (F).
Clicking on the next two images brings up movies recorded live while a user
interactively manipulated our real-time light field
viewer. The input to the viewer in each case is a single light field
micrograph. The rightmost image brings up a movie also recorded live while a
user interactively manipulated our light field viewer, but in this case the
viewer is playing back a light field video. The video was recorded from the
specimen at 16 frames per second with 2 x 2 pixel binning. Each frame in this
video is a complete 4D light field, recorded on the Retiga's sensor as a 2D
array of circular subimages. Hence, this is a 5D dataset. As a result, while
playing back the video the user has full control over viewing direction and
focus. Indeed, near the end of the movie you can see the user manipulating the
viewpoint interactively.
The manipulation of viewpoint in this last movie shows an advantage of light
field microscopy over scanning microscopes for capturing transient events. On
each frame of the original video, an entire light field was captured, not a
single view. From each light field, a focal stack or volume can be
reconstructed as described in our
SIGGRAPH 2006 paper.
Hence, we can construct a time-varying volume dataset. Each frame in such a
dataset would represent the 3D structure of the transient event at a single
instant in time, without the mid-frame shearing present in data captured
with scanning confocal microscopes.
© 2009
Marc Levoy
Last update:
July 13, 2009 01:05:54 PM










{kind=link}
{kind=link}